
Quantifying structural relationships of metal-binding sites suggests origins of biological electron transfer
Motivated by how crucial redox reactions are for life, the authors take a look at how early in molecular evolution catalysis of such reactions has emerged. They found a couple of small motifs, binding metals or metal-associated ligands, of similar geometry and sequence used across all proteins. That may be seen as indicator that peptides with redox functionality could have existed before first proteins. At the core of the study, a new structure-annotated homology, ligand-extended (sahle) score is used to calculate a distance between structural fragments. After reducing fragment space to representatives, the distance network is transformed into a minimum spanning tree (MSP) and enriched with genomic data to get phylogeny attached to the fragments. This leads to a small set of central motifs which serve as origin for the rest of the MSP. Verifying their central motifs with other studies leads the authors to the assumption that peptides carrying those motifs may have existed more than 3.8 billion years ago.
Folding the unfoldable: Using AlphaFold to explore spurious proteins
The authors apply AlphaFold2 to AntiFam, which contains 250 sequence profiles annotated as “spurious” proteins, either because they are believed to be spurious translations, they overlap with known non-coding RNA genes, or because they are found on the opposite strand of known genes. Their first result is that there is a strong correlation between the mean plDDT and the length of the sequence, with shorter sequences showing higher mean plDDT both with the AntiFam sequences and with randomly generated amino acid sequences. But, ignoring sequences <100 residues, the mean plDDT scores are higher for SwissProt proteins compared to AntiFam spurious proteins. This proved false for only one AntiFam entry, and on closer inspection it looks like there’s a lot of evidence for this entry representing real functional proteins - so the authors deposited this entry back into Pfam. Overall a nice analysis showing that AlphaFold2 can provide an additional quality control estimate for spurious protein profiles, with the caveat that it shouldn’t be used for short sequence lengths.
Can AlphaFold2 predict the impact of missense mutations on structure?
No, it can not. The authors show three examples in which AlphaFold2 fails in predicting the ‘structure-disrupted’ protein successfully. This is not that surprising considering the fact that AlphaFold is trained mostly on wild type structures and it is supposed to be resistant to noise. However they offer an interesting showcase analyzing those examples in detail.
Figure of the Week
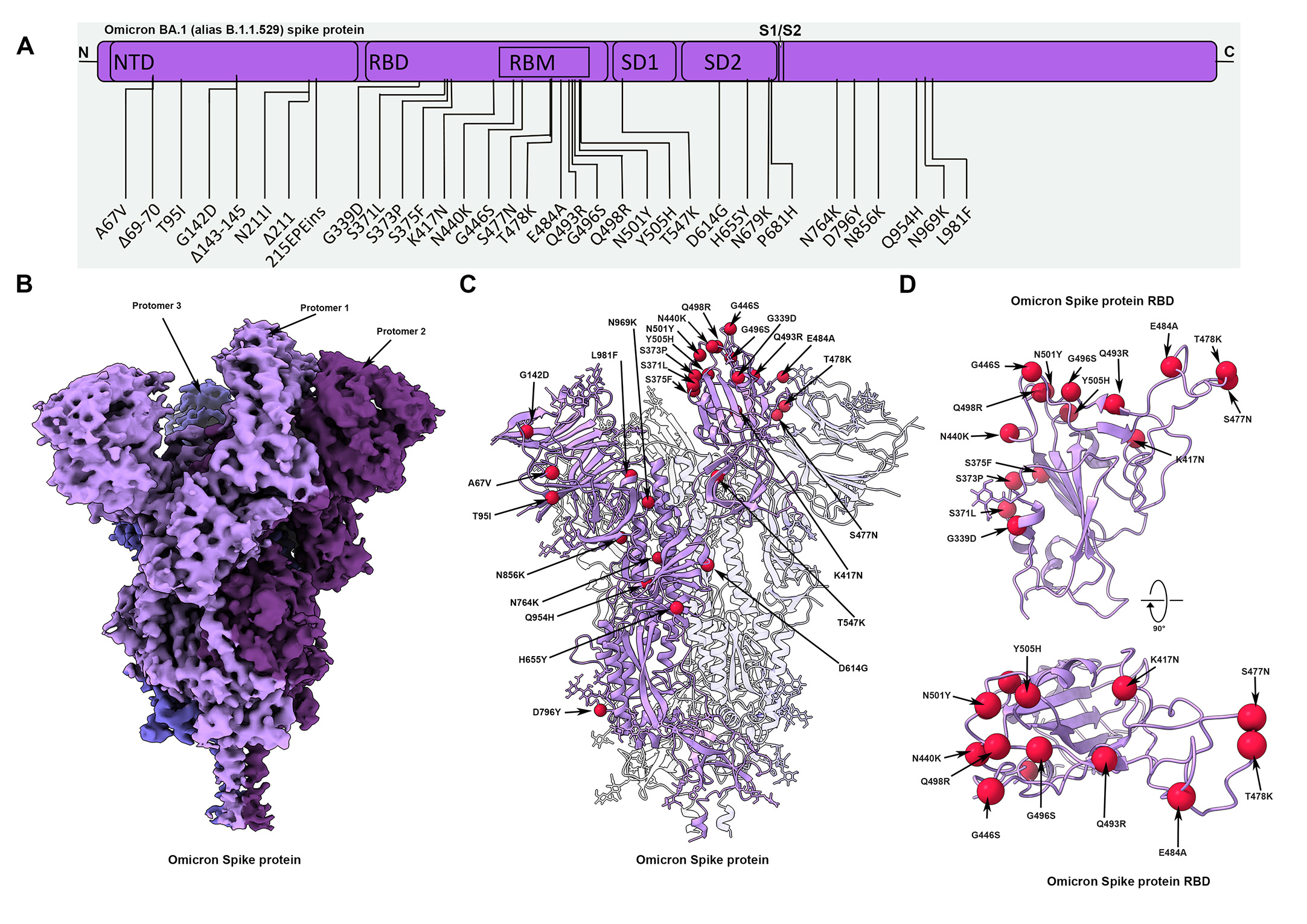
Fig. 1. Cryo-EM structure of the Omicron spike protein. (A) A schematic diagram illustrating the domain arrangement of the spike protein. Mutations present in the Omicron variant spike protein are labeled. (B) Cryo-EM map of the Omicron spike protein at 2.79 Å resolution. Protomers are colored in different shades of purple. (C) Cryo-EM structure of Omicron spike protein indicating the locations of modeled mutations on one protomer. (D) The Omicron spike receptor-binding domain (RBD) shown in two orthogonal orientations with Cα positions of the mutated residues shown as red spheres.
Source: SARS-CoV-2 Omicron variant: Antibody evasion and cryo-EM structure of spike protein–ACE2 complex
Some more
- Reconfigurable asymmetric protein assemblies through implicit negative design
- Soft disorder modulates the assembly path of protein complexes
- SARS-CoV-2 Omicron variant: Antibody evasion and cryo-EM structure of spike protein–ACE2 complex
- Evolving cellular automata schemes for protein folding modeling using the Rosetta atomic representation
- The accuracy of protein structures in solution determined by AlphaFold and NMR
- Ins and outs of AlphaFold2 transmembrane protein structure predictions
- Single-sequence protein structure prediction using supervised transformer protein language models
- This Guardian article about black boxes in science
- A fun twitter thread on pretty protein structures:
Protein twitter! What is the most aesthetically pleasing structure? I am making new desktop wallpapers for the beamlines...
— Dr Eleanor Campbell 🦑 (@Elamanor) January 17, 2022