Fundamental behaviors emerge from simulations of a living minimal cell
Can we simulate a full living cell with all its chemicals while taking into account spatial heterogeneity? The authors describe a significant step in that direction by simulating a living minimal cell (JCVI-syn3A) with over 7000 reactions governing the evolution of its metabolites, proteins, mRNAs, tRNAs, DNA and ribosomes. A coarse grained spatially resolved model (at 4 nm resolution) was initialised based on cryo-ET images providing the cell morphology and the location of the ribosomes while other macromolecules were added to it. Simulations were run with a hybrid stochastic and deterministic setup both on the spatially resolved model and on a simpler well stirred model without spatial heterogeneity. The latter was necessary as the computational cost for the spatial model was too high and growth of the cell could not properly be accounted for yet. Future work should address these limitations, but the current model was already able to showcase interesting emerging behaviours which fit well with experimental observations while providing additional time-resolved details. For instance, the authors could measure distributions of mRNA half-lives and protein production, when and where ATP was used, connections between metabolism and cell growth, and DNA replication events.
Petabase-scale sequence alignment catalyses viral discovery
The authors perform a high throughput alignment-based sequence search across over 10 petabytes of sequence read data with the purpose of identifying new RNA viruses. Their approach has a higher sensitivity than k-mer index approaches, while being much faster than de novo assembly approaches. The viral search is underpinned by a “palmprint” - a set of three essential motifs in RNA-dependent RNA polymerases which form its catalytic core. Novel palmprints identified across the huge database increased the number of known RNA viruses nearly ten-fold. The authors then focus on three relevant viral families - Coronoviridae (of SARS-CoV-2 fame), delta virus (such as the one causing viral hepatitis), and huge phages from microbiome research. The polished and comprehensive Serratus website provides access to all these analyses and the raw data. You can even find matches for your own virus of interest.
Cellular homologs of the double jelly-roll major capsid proteins clarify the origins of an ancient virus kingdom
Viruses are a distinct group of biological entitites and their origin is a hotly debated problem. While most core components of most viral lineages seem to precede the last universal common ancestor (LUCA), there are indications that, within various structural components, different capsid proteins may have evolved from the recruitment of diverse cellular proteins, especially those involved in the metabolism of carbohydrates. In this study, the authors investigate the origins of the major capsid protein of a broad variety of double-stranded DNA viruses, a protein long thought to represent a viral innovation. Using protein structure comparison, they traced this protein family to a bacterial cellular ancestor likely involved in carbohydrate metabolism, rephrasing the history of these viruses while reinforcing the chimeric view on virus origins.
Figure of the Week
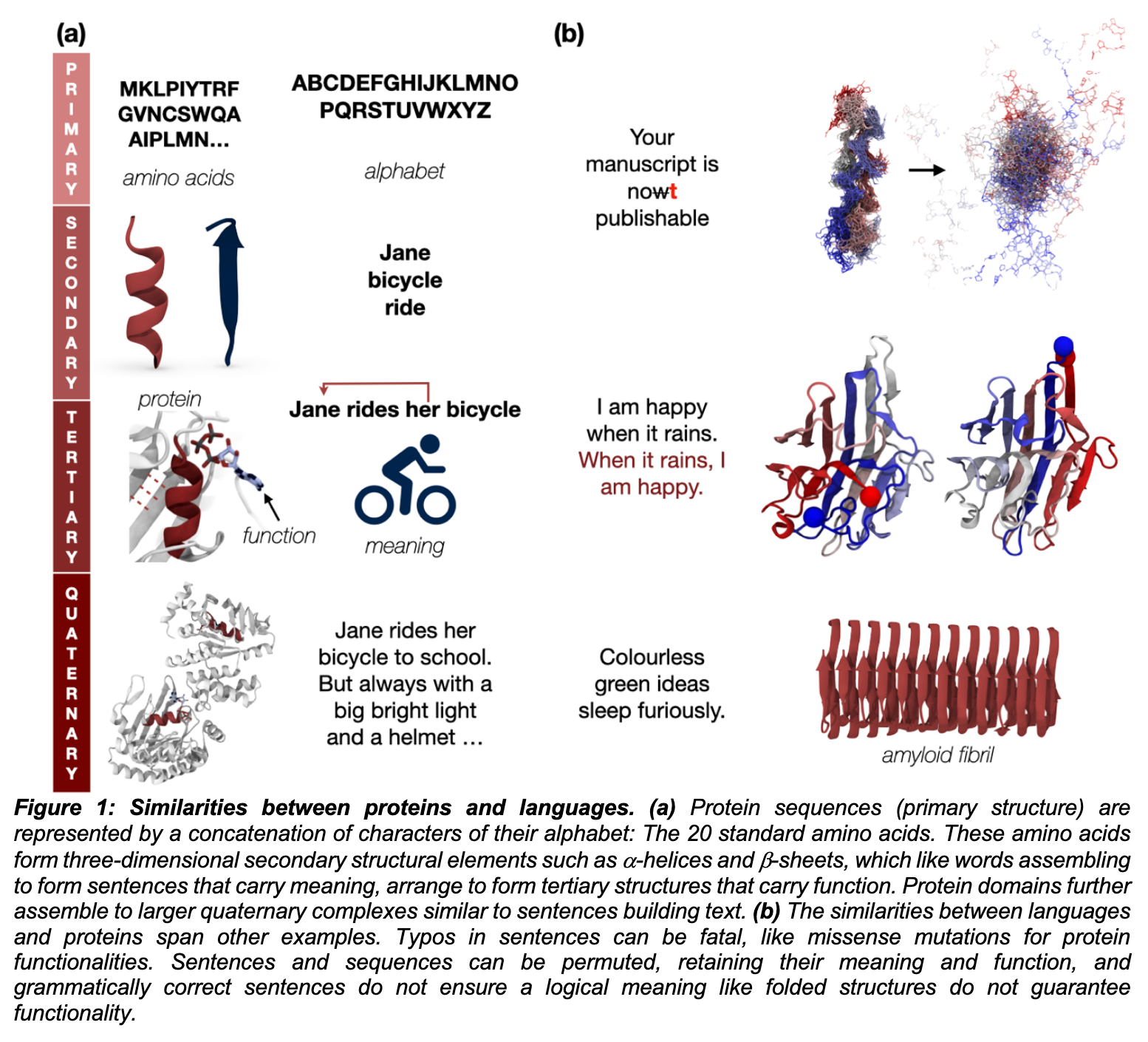
Source: Towards Controllable Protein design with Conditional Transformers
Some more
- AlphaFold Accelerates Artificial Intelligence Powered Drug Discovery: Efficient Discovery of a Novel Cyclin-dependent Kinase 20 (CDK20) Small Molecule Inhibitor
- Proteome-scale mapping of binding sites in the unstructured regions of the human proteome
- Fragment Hotspot Mapping to Identify Selectivity-Determining Regions between Related Proteins
- The computational models of AlphaFold2 and RoseTTAfold carry protein foldability information
- Design in the DARK: Learning Deep Generative Models for De Novo Protein Design
- Enhancing protein inter-residue real distance prediction by scrutinising deep learning models
- Label-free visual proteomics: Coupling MS- and EM-based approaches in structural biology
- Towards Controllable Protein design with Conditional Transformers
- Lira: Rotational Invariant Shape and Electrostatic Descriptors for Small Molecules and Protein Pockets based on Real Spherical Harmonics
- ATLIGATOR: Editing protein interactions with an atlas-based approach
- The AlphaFold Database now has over a million structures
On Neglected Tropical Disease Day, we teamed up with @DeepMind to publish 190,000+ relevant protein structure predictions in the #AlphaFold database – freely available to all! #WorldNTDDay #beatNTDhttps://t.co/Mvq4cC2ilI pic.twitter.com/IRUXwq6pVg
— EMBL-EBI (@emblebi) January 28, 2022