
Evolutionary velocity with protein language models predicts evolutionary dynamics of diverse proteins
Protein language models are revolutionizing protein bioinformatics. From the prediction of protein sequence features and the classification of protein families, to important contributions to protein structure prediction. In this paper, Hie et al go a step further and use them to model protein sequence evolution as a “walk through the protein sequence landscape”. Based on the ESM-1b protein language model, which the authors say learnt general protein evolutionary patterns, they developed an approach that used distances in embedded space to build a sequence similarity network on top of which they map a “vector field” that translates into the evolutionary dynamics within the sequences analysed. The approach starts by the computation of the sequence embeddings for a target set of sequences using the language model. KNN as implemented in UMAP is then used to find the neighbours of each sequence in embedded space, mimicking a sequence similarity network. Then, the change in the language model pseudo-likelihood in each edge is used to assign a “velocity” vector whose direction represents an evolutionary transition. While it is not clear to me why this last step has the evolutionary meaning claimed, the authors demonstrate that for different protein families the internal structure of resulting network and the vector field generated captures their known evolutionary patterns. What makes this work even more exciting is that the method is made available as a python package (evolocity), which relies on Scanpy and anndata, two other packages that facilitate the analysis of large multidimensional annotated data. The package comes with multiple tutorials that can be run in Google Colab; this makes it easier to explore it for our favourite sets of sequences. But be warned: you may run into crashes due to memory overload in Colab. Nevertheless, it is exciting that such a tool is now made easily available and, while there is some amount of flexibility within the package, the expansion to other clustering methods and further language models would make it even more rich.
Scalable Fragment-Based 3D Molecular Design with Reinforcement Learning
The authors describe an interesting molecule design method that combines Markov decision processes with machine learning. It consists in a hierarchical agent that sequentially places molecular fragments with predefined shapes in 3D space. They compare this methodology with the intuition of an organic chemist who, when faced with a task to modify a molecule, would think of it in terms of adding new fragments and functional groups rather than single atoms. This method allows building molecules with more than 100 atoms, a task that other approaches that take the 3D structure into consideration usually fail to accomplish. Moreover, they suggest that it is possible to optimize different properties during the design process, like hydrophilicity or the affinity with a specific pocket. This makes this method extremely versatile, leading to many potential applications such as: small molecule and peptide drug design or design of ligands and organocatalysts.
Molecular electronics sensors on a scalable semiconductor chip: A platform for single-molecule measurement of binding kinetics and enzyme activity
The future is now! The authors realise the 70s' dream of using single molecules as circuit elements, with a CMOS-integrated circuit chip consisting of an array of nanoelectrode-based single-molecule sensors “bridged” by a molecular wire between the nanoelectrodes. The biomolecule of interest is conjugated to the bridge molecule to act as a probe (a process that takes 10 seconds), the target molecule is added into the equation, and then the current through the bridge is monitored - if the target molecule interacts with the probe then this can be seen via the current pulses. And not just seen either - Hidden Markov models are used to analyse the current signals, with bound and unbound being the hidden states, and by comparing the fraction of bound vs unbound across different target concentrations they obtain pretty accurate estimates for the binding affinity $K_d$ as well! The authors demonstrate an impressive variety of applications - DNA hybridization of complementary oligos (including determining the melting temperature of the DNA duplex, and showing sensitivity to introduced mismatches), protein and small-molecule binding, DNA aptamer-target binding (shown using aptamers designed to bind SARS-CoV2 proteins), and enzyme activity (shown using a CRISPR/Cas system with the guide RNA targeting the Spike protein gene, and by distinguishing between DNA polymerase enzyme incorporation events) among others. They even show that adding a bunch of saliva or other highly complex contaminants doesn’t mess up sensing, meaning it’s perfect for diagnostic tests. Overall, the chip is sensitive and specific, can be multiplexed to target many kinds of interactions and molecules, highly scalable way past Moore’s law, fast (seconds) and cheap (~1$). Looks like a bright future for obtaining high quality drug-target binding affinity data to train our methods!
Figure of the Week
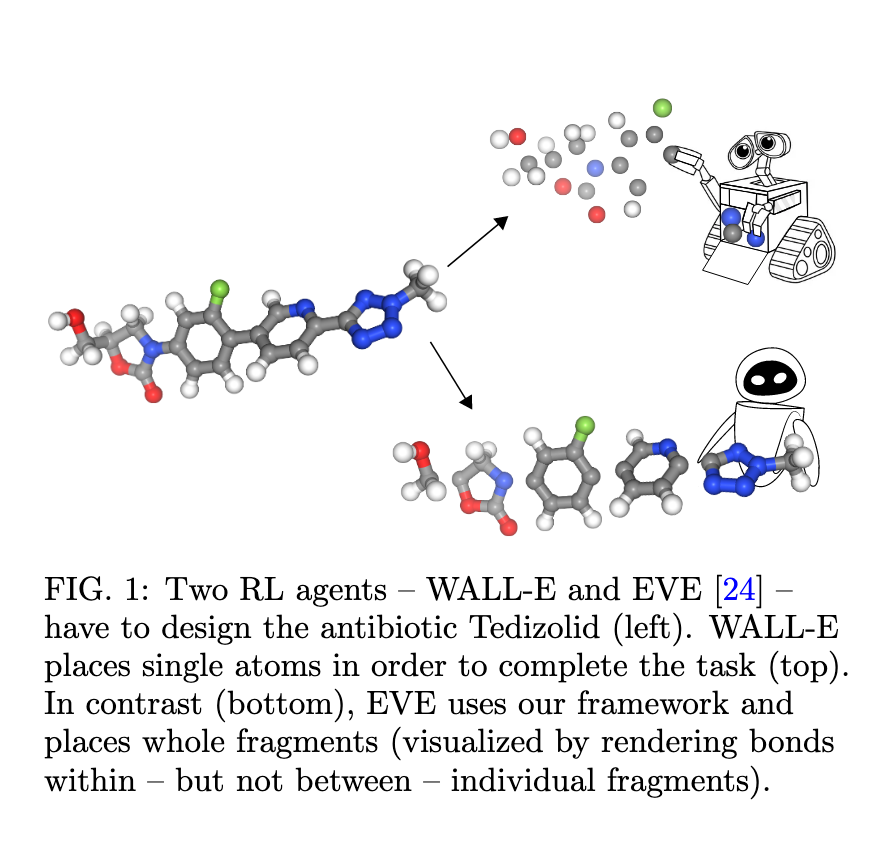
Source: Scalable Fragment-Based 3D Molecular Design with Reinforcement Learning
Some more
- Cryo-EM and artificial intelligence visualize endogenous protein community members
- What stabilizes pre-folded structures in the intrinsically disordered α-helical binding motifs?
- Riemannian Geometry and Molecular Surfaces I: Spectrum of the Laplacian
- Computational Modeling of Molecular Structures Guided by Hydrogen-Exchange Data
- Current structure predictors are not learning the physics of protein folding
- AlphaDesign: A graph protein design method and benchmark on AlphaFoldDB
- DisEnrich: Database of Enriched Regions in Human Dark Proteome
- EViS: An Enhanced Virtual Screening Approach Based on Pocket–Ligand Similarity
- Assessment of AlphaFold2 residue conformations for human proteins
- ChemPlot, a Python library for chemical space visualization
- SPOT-Contact-LM: Improving Single-Sequence-Based Prediction of Protein Contact Map using a Transformer Language Model
- A Springer book on Artificial Intelligence in Drug Design
- UniProt has a Machine Learning Challenge aimed at creating computational methods to predict metal binding sites across the whole of UniProtKB
- Some resources on the latest hot topic, protein design via hallucination: A video tutorial and ColabFold support
Adding support for binder hallucination if anyone wants to try! (Code is very experimental, not intended for practical use... only use for art/science) 😀https://t.co/OOPp8kSu2Z pic.twitter.com/WUu9LGKIwp
— Sergey Ovchinnikov 🇺🇦 (@sokrypton) February 3, 2022