
Foldseek: fast and accurate protein structure search
In the era of high-throuhput, high accuracy protein structure prediction, accurate and fast protein structure search and alignment methods are essential. They can be used to annotate folds, find remote homologous relationships undetected at the sequence level, or for fold classification. In this paper, van Kempen et al. describe Foldseek, the MMseqs2 of protein structure alignment. The method is based on a novel type of structural alphabet with 20 states that describes for each residue i the geometric conformation with its spatially closest residue j, thus differentiating it from structural alphabets that don’t consider spatial neighbours. Structures are compared based on their “sequence” in this alphabet using the MMseqs2 approach, and then aligned locally or globally. Foldseek is as sensitive as TMalign and almost as sensitive as DALI, but it is considerably faster! As an example, the authors show that Foldseek does in 5 seconds what would take TMalign 33 hours and Dali 10 days. The online availability of Foldseek as a server, allows users to query their favourite structure against the AlphaFold database or the PDB, and the easy installation process allows for extremely fast many-against-many sensitive structure comparisons.
Conformational ensembles of intrinsically disordered proteins and flexible multidomain proteins
A comprehensive review on generating integrated conformational ensembles for proteins with different levels of conformational heterogeneity. The authors start off by describing typical steps in ensemble determination: computational conformation sampling, experimental methods to determine structure, forward models to calculate experimental observables from the ensemble, and finally refining the ensemble based on the experimental output. They then go into detail on a few examples of multidomain protein and protein-nucleic acid complexes, lipid membranes, multimodal ensembles with different subpopulations, and small-molecule binding. The authors also discuss some interesting future opportunities - improving force fields and forward models, using computational structure prediction methods instead of experimental methods now that they are more comparable, and looking more into different time scales and heterogenous mixtures of proteins.
EquiBind: Geometric Deep Learning for Drug Binding Structure Prediction
In EquiDock, the authors introduced a new graph neural network approach for rigid protein-protein docking, by training the network to output the Euclidean SE(3) transformation (i.e the rotation and translation) required to move one protein (labelled the “ligand”) with respect to the other (labelled the receptor). This approach had a couple of neat features: it’s SE(3)-equivariant, meaning it returns the same docked complex irrespective of the inital orientations and positions of the input proteins; it uses a k-NN protein graph as input with SE(3)-invariant edge features based on local coordinate frames, the same approach used by AlphaFold; it uses multi-headed attention to find “keypoints” for each protein, with learning ensuring that these represent binding pockets, and then optimally aligns the “ligand” protein to match these keypoints; and it’s very very fast. Now EquiBind extends this approach to solve small molecule docking instead, with a key addition - the ligand, which is now a small molecule represented in a similar manner as the protein graph, is flexible! This is acheived by learning deformations of the atom point cloud and then rotating torsion angles in the ligand to best match the deformed point cloud. EquiBind takes mere seconds, perfect for virtual screening. Some opportunities for improvement - explicitly modeling residue sidechains, allowing for receptor flexibility, and handling multi-ligand docking.
Here’s a twitter thread with more info:
New paper! Fast blind structural drug binding using geometry&deep learning.
— Octavian Ganea (@octavianEganea) February 11, 2022
Understanding how a small drug-like molecule attaches to a target protein is a core problem in drug discovery. By interacting with specific surface areas, drugs can change proteins' functions. 1/ pic.twitter.com/6N1CAMOQrd
Figure of the Week
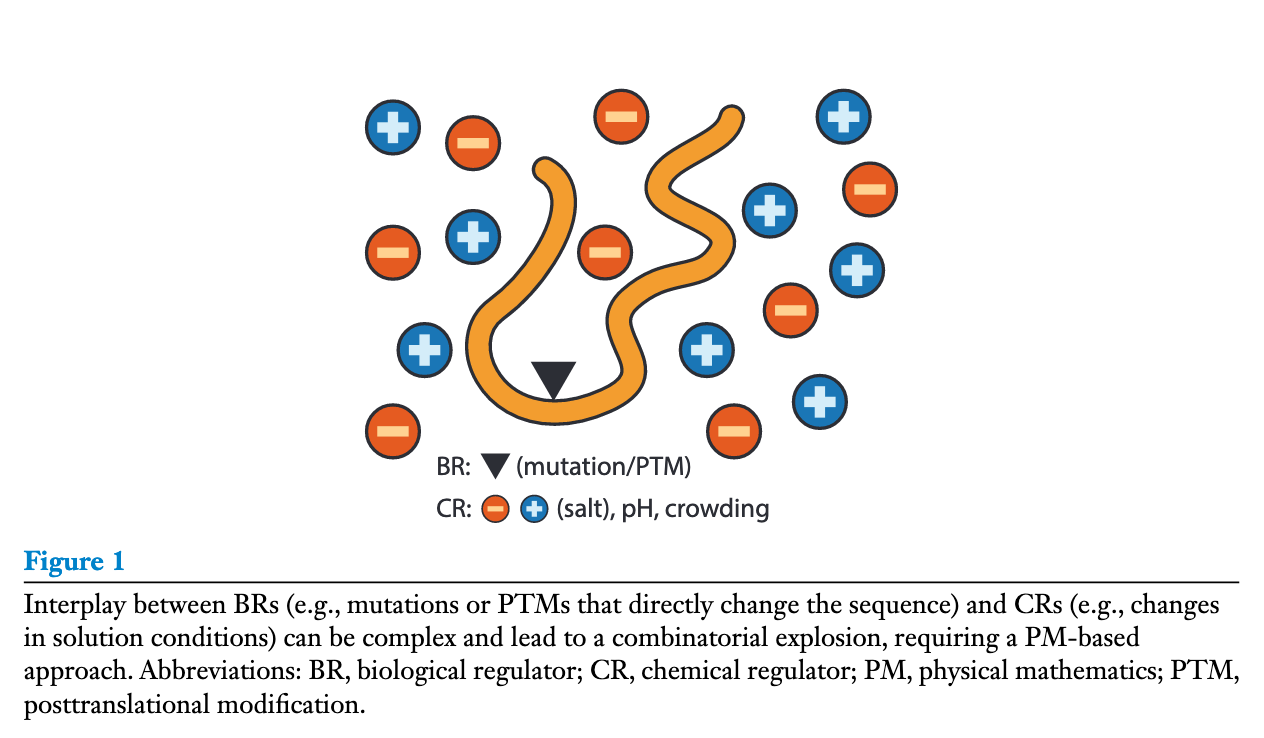
Source: Rules of Physical Mathematics Govern Intrinsically Disordered Proteins
Some more
- Protein sequence design with a learned potential
- Rules of Physical Mathematics Govern Intrinsically Disordered Proteins
- A backbone-centred energy function of neural networks for protein design
- Using machine-learning-driven approaches to boost hot-spot’s knowledge
- Structures of the Omicron Spike trimer with ACE2 and an anti-Omicron antibody
- PirePred: An Accurate Online Consensus Tool to Interpret Newborn Screening–Related Genetic Variants in Structural Context
- A beautiful Blender tutorial on looking inside a protein surface
- PDBeKB now shows ligands and cofactors “transplanted” into AlphaFold models, courtesy of AlphaFill which we’ve talked about before
#AlphaFill is now available in #PDBeKB thanks to #3Dbeacons. https://t.co/J6v0wHtHXR
— Robbie Joosten (@Robbie_Joosten) February 10, 2022