
A double issue covering the last two weeks of updates in structural bioinformatics
Using deep learning to annotate the protein universe
Each year the wealth of protein-coding sequences uncovered by multiple genomic projects increases exponentially. This makes their annotation for functional features challenging but also more and more important. Metagenomic projects are an especially big source of novel protein sequences and while many for sure represent families completly new to us, a much bigger fraction most likely correspond to highly divergent forms of well known families that just escape detection by HMM-based annotation methods. In this work, the authors trained a network on the domain families in Pfam and demonstrate how deep learning allows the annotation and classification of sequences seemingly without clear homologs, expanding the annotation coverage of the protein universe. The method is useful for the annotation of single domain sequences and allowed for the merging of DUF families with others well known. It does not, however, map annotations into the full-length sequence. The authors circumvent the annotation of multi-domain sequences without known domain boundaries by using a sliding window. The code is freely available and a Colab notebook makes it really convenient to play with your favourite protein. One word of caution however: while powerful and indeed superior in sensitivity to HMMs, the network always provides a result; it always finds the closest annotation in embeded space for a given sequence, even if it is really far away.
In silico evolution of nucleic acid-binding proteins from a nonfunctional scaffold
The authors tackle the fascinating problem of designing proteins with a given function, using simulated directed evolution. To make the task even more challenging they demonstrate their approach, Proseeker, on a completely de novo designed repeat protein with no existing function, designed just to optimize its consecutive alpha-helix folding capabilities. To imbue this protein with nucleotide-binding functionality, the authors create an assessment library of phycochemical descriptors describing short peptides from pentatricopeptide repeat proteins, specifically those covering the RNA-binding residues. These descriptors are clustered and any newly evolved protein is compared to the representative descriptors from each cluster to determine its fitness. Two residues are mutated at each step in the process, with residues around the first mutation site getting a small boost in being selected as the second mutation site, so as to boost creation of mutational islands and epistatic interactions. Then, selection is done computationally using DP-Bind - an SVM trained to predict DNA binding ability. The top scoring 10% variants are used to generate the next round of mutants. Proseeker is experimentally validated by producing five synthetically evolved proteins and running multiple in vitro and in vivo (with a pretty neat new design) assays, showing that three of them actually do have nucleotide-binding activity, an amazing result. Another nice result of the paper is describing all the different variables that are important for getting this kind of approach to work, and systematically varying each one to show its effect - of course this is only possible since the evolution is in silico!
PyUUL provides an interface between biological structures and deep learning algorithms
For the structural biologists who were not born with Machine Learning or fancy Neural Networks (NN), using these new methods can be sometimes intimidating. pyUUL is one of those tools that will help sand down some of the rough edges and make you finally jump on the bandwagon like the other cool kids. The aim of this package is to transform any 3D structures of your choice (protein, small molecules or nucleic acids) into a meaningful representations (voxel-based, surface or volumetric point-cloud) directly exploitable by any NN architecture. At the end, this idea is simple, but nonetheless very powerful. The authors also present multiple test cases, ranging from alpha helix identification, structure-based clustering and small-molecule optimization. Based on PyTorch, all the operations can be run on both CPU and GPU. The package is available here and more importantly it comes with documentation describing some of the examples presented in the papers.
Systematic evaluation of computational tools to predict the effects of mutations on protein stability in the absence of experimental structures
Methods for the prediction of protein stability upon mutation are usually evaluated and developed with experimental structures in mind. With the vast majority of structures for proteins being models and predictions, the authors tackled the question of how common mutation stability prediction tools would perform on protein structure models. They evaluated the performance of 14 protein stability predictors on a self-created data set of models based on a curated subset of structures in the ProTherm data set using MODELLER and AlphaFold2. The authors found that the protein stability predictions deteriorate for homology models with a target-template identity below 40%; especially for machine learning and structure based methods. The predictions on AlphaFold2 models were mostly consistent with predictions on experimental structures, which opens up stability predictions with the same accuracy as in experimental structures to all proteins. A limitation we see here is the entanglement of the evaluation of the models of proteins in the ProTherm data base, which is usually also used for the development of stability prediction methods.
Exploring protein-protein interactions at the proteome level
The biological activity of proteins is defined by how they interact with other biological molecules, from small ligands, to nucleic acids, sugars and other proteins. Usually proteins do not act by themselves, and form strong or transient complexes with other proteins, building large macromolecular machines or transient interaction networks. It is thus essential to not only understand the three-dimensional structure of individual proteins but also in complex with their interaction partners. In this review, Elhabashy et al. discuss the different approaches available to analyse, predict and model such interactions at a large-scale. From experimental methods, to computational and hybrid approaches, they provide a concise but extremely informative and insightful state-of-the-art for the modelling of protein-protein interactions at the proteome level, also highlighting the recent role of machine learning in such tasks.
Figure(s) of the Week(s)
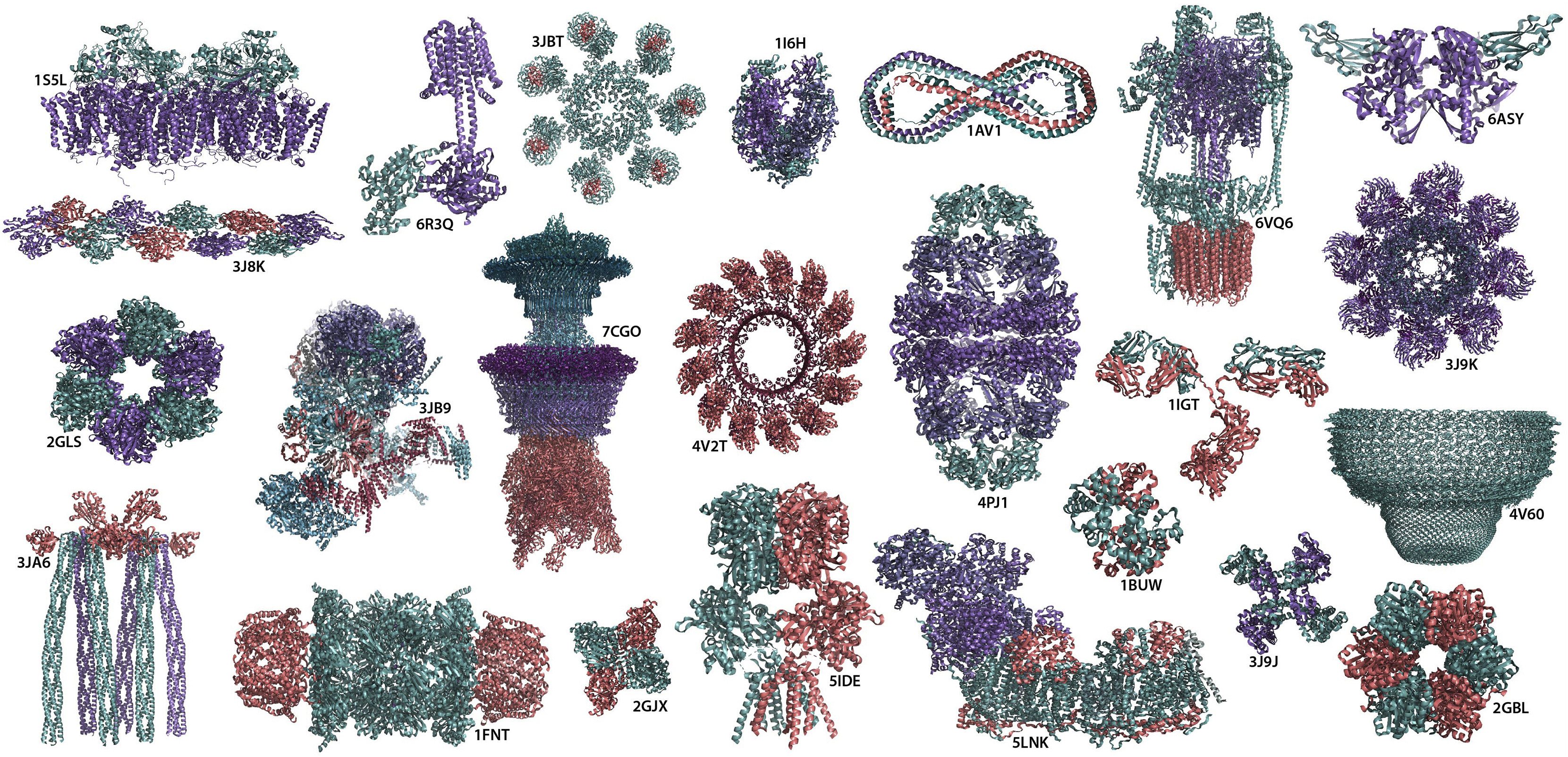
Figure 1. Molecular machinery of life Gallery of supramolecular complexes formed through protein-protein interactions. PDB IDs of shown structures from the left to right are as follows 1S5L: photosynthetic oxygen evolving center; 3J8K: tilted state of actin T2; 2GLS: glutamine synthetase; 3JA6: novel kinase conformational switch in bacterial chemotaxis signaling; 6R3Q: membrane adenylyl cyclase bound to an activated stimulatory G protein; 3JB9: yeast spliceosome; 1FNT: 20s proteasome from yeast in complex with the proteasome activator pa26 from Trypanosoma brucei; 3JBT: the Apaf-1 apoptosome; 7CGO: the flagellar motor-hook complex from Salmonella; 2GJX: human β-hexosaminidase A; 1I6H: RNA polymerase II elongation complex; 4V2T: membrane-embedded pleurotolysin pore with 13-fold symmetry; 5IDE: GluA2/3 AMPA receptor heterotetramer (model I); 1AV1: human apolipoprotein A-I; 4PJ1: human mitochondrial chaperonin symmetrical ‘football’ complex; 5LNK: ovine respiratory complex I; 6VQ6: mammalian V-ATPase from rat brain; 1IGT: immunoglobulin; 1BUW: s-nitroso-nitrosyl human hemoglobin A; 6ASY: human endoplasmic reticulum chaperone BiP; 3J9K: dark apoptosome in complex with the Dronc CARD domain; 4V60: rat liver half-vault; 3J9J: capsaicin receptor TRPV1; and 2GBL: circadian clock protein KaiC
Source: Exploring protein-protein interactions at the proteome level
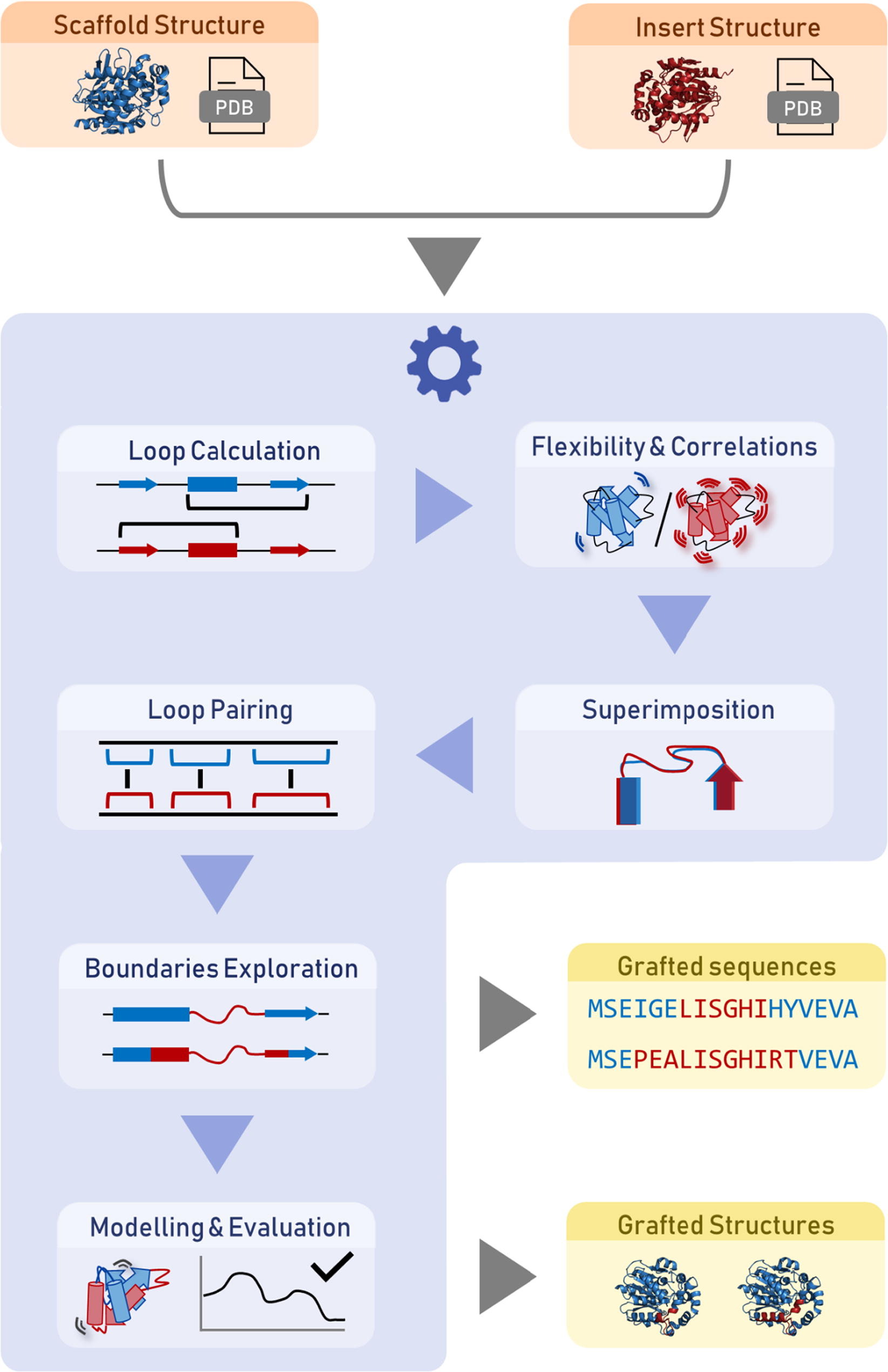
Fig. 3. Illustration of the LoopGrafter workflow. The server starts with the input of the PDB files or codes of two structures (orange): (i) the loop receptor (scaffold) and (ii) the loop donator (insert). The tool then calculates the secondary structures of all the loops, assesses the protein flexibility using normal mode analysis, and performs a superimposition of the two structures (blue). Once the user selects the loops of interest, the loops are paired. The suitable loop boundaries are then explored based on geometric restrictions so that maximal sequence diversity is probed in the designed grafted variants. The possible solutions are collected as a list of sequences, and the structures are then modeled and ranked by the respective stability scores obtained from Rosetta and MODELLER (yellow). The web service is provided free of charge for non-commercial use: https://loschmidt.chemi.muni.cz/loopgrafter/.
Source: Tools for computational design and high-throughput screening of therapeutic enzymes
Many more
- DisProt in 2022: improved quality and accessibility of protein intrinsic disorder annotation
- Accurate self-assessment in 2D: protein contact map quality estimation by deep evolutionary reconciliation
- Application of Homology Modeling by Enhanced Profile–Profile Alignment and Flexible-Fitting Simulation to Cryo-EM Based Structure Determination
- ProteinBERT: a universal deep-learning model of protein sequence and function
- MeCOM: A Method for Comparing Three-Dimensional Metalloenzyme Active Sites
- Model agnostic generation of counterfactual explanations for molecules
- Molecular modeling in drug discovery
- Ancestral reconstruction of duplicated signaling proteins reveals the evolution of signaling specificity
- Tools for computational design and high-throughput screening of therapeutic enzymes
- Visualizing protein breathing motions associated with aromatic ring flipping
- Forty years in cryoEM of membrane proteins
- The clinical variant interpretation portal DECIPHER now has AlphaFold structures:
DECIPHER portal for clinical variant interpretation (free-to-use) now contains AlphaFold structures, annotated with known pathogenic variants, displayed interactively. https://t.co/rgbBXB5T4M pic.twitter.com/LLKmWs4Clf
— Matt Hurles (@mehurles) February 18, 2022
- Structural basis for SARS-CoV-2 Delta variant recognition of ACE2 receptor and broadly neutralizing antibodies
- SWAAT Bioinformatics Workflow for Protein Structure-Based Annotation of ADME Gene Variants
- A Step-by-step Guide on How to Construct quasi-Markov State Models to Study Functional Conformational Changes of Biological Macromolecules
- Modeling of protein conformational changes with Rosetta guided by limited experimental data
- TorchDrug: A Powerful and Flexible Machine Learning Platform for Drug Discovery
- Systematic identification of conditionally folded intrinsically disordered regions by AlphaFold2
- A Colab notebook to find protein binding sites on your structure with dMASIF
We at LPDI are happy to announce that dMaSIF site is now available on Google Colab thanks to @hamed_khakzad @rneschneuing @befcorreia. It has never been easier to visually identify protein binding sites based on surface features!
— Casper Goverde (@CasperGoverde) February 21, 2022
Try it here: https://t.co/xCcEBkkKBd pic.twitter.com/LadZsJ3PFm
- TorsionNet: A Deep Neural Network to Rapidly Predict Small-Molecule Torsional Energy Profiles with the Accuracy of Quantum Mechanics
- Localpdb— a Python package to manage protein structures and their annotations
- The structural context of PTMs at a proteome wide scale
- From evolution to folding of repeat proteins
- Protein-protein interaction prediction for targeted protein degradation
- SiteMotif: A graph-based algorithm for deriving structural motifs in Protein Ligand binding sites
- PSnpBind: a database of mutated binding site protein–ligand complexes constructed using a multithreaded virtual screening workflow