
A bundle of protein interactomes
This week, three different papers addressing the 3-dimensional modelling of protein interactions at the cellular level were released, all paving the way to the automated and high-accuracy modelling of entire cells in 3D. The first, Computed structures of core eukaryotic protein complexes, addresses the problem of predicting the composition and accurately modelling the 3D structure of protein macromolecular assemblies at the same time. The combination of RoseTTAFold for pair prediction with AlphaFold2 for structure modelling allowed for the construction of more than 1000 protein-protein assemblies from baker’s yeast. This not only included previously known interactions, but also more than 100 unknown others that may highlight previously unknown functions.
In the second paper, Towards a structurally resolved human protein interaction network, the authors use AlphaFold2 to model the 3-dimensional structure of more than 60’000 previously known protein interactions in the human proteome, providing building blocks to the construction of larger assemblies.
Both works pave the way to the modelling of the interaction networks and high-accuracy simulation of cells in 3D; something that can be now more easily done, as highlighted by the third paper, Building structural models of a whole mycoplasma cell. In this work, the authors used experimental and homology-modelled macromolecular structures and lattice-based models of a genome to generate the first 3D whole cell model of a eukaryote, Mycoplasma genitalium.
If we thought one year ago that the structural biology field was changed forever due to the onset of deep learning, advancements in computer graphics and whole cell simulations together with this boosting revolution will definitely change the way we study (molecular) biology.
Mining for encrypted peptide antibiotics in the human proteome
Using their previously developed predictor of antimicrobial efficacy for antimicrobial peptides (AMP), the authors of this paper identified 2603 AMPs encoded in the human proteome. A selection of 55 representative peptides show the capability to not only kill bacteria by targeting their membrane, they also do not readily select for antibiotic resistance and show anti-infective activity in mouse models. It is quite astonishing that a search algorithm based only on 3(!) physicochemical properties (net charge, hydrophobicity and length) was enough to find new promising candidates which were shown to be viable antibiotics. Here’s a blog post about it too.
Iterative Refinement Graph Neural Network for Antibody Sequence-Structure Co-design
A fascinating protein design task, namely the joint design of an antibody sequence and 3D structure. The authors formulate this as a graph-generation problem and combine autoregressive models with iterative refinement to generate a full graph with node labels, edge labels and coordinates. In each generation step, the model learns to revise a current antibody graph and predict the label of the next residue. There’s one message passing network to predict the amino acid, and then another one to predict the new coordinates of all residues given the newly added amino acid. Unlike AlphaFold, this approach predicts the structure of an incomplete sequence without a specified MSA. This allows them to optimize the sequence-structure combo according to specific requirements. For instance, one of the validation experiments aims to identify neutralizing antibodies for COVID-19 with sequence constraints such as low net charge, no glycosylation motifs, and no repeating stretches of amino acids. This level of control could be very useful for other protein design tasks as well.
Figure of the Week
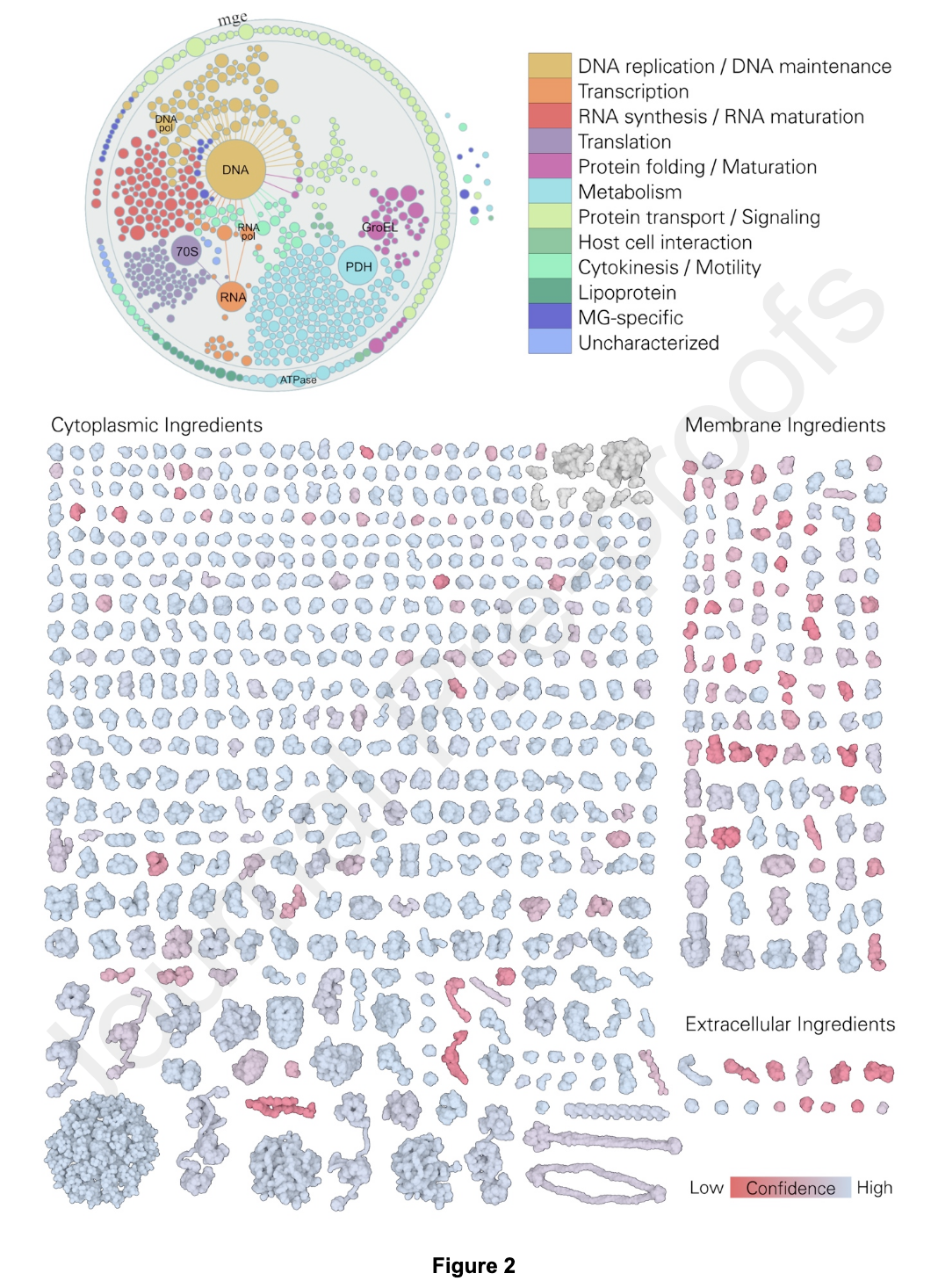
Figure 2. 3D-WC-MG recipe. Top, graphical visualization of the proteome of MG, separated and colored by function, displayed with
Mesoscope, our tool for integration and curation of mesoscale data (presented in more detail in Figure 10). Bottom, reconstructed proteome of MG displayed with atom radius scaled by a factor of three and colored by structure confidence score; RNA molecules are colored in grey.
Source: Building Structural Models of a Whole Mycoplasma Cell
Some more
- Structure-aware generation of drug-like molecules
- Ligand binding remodels protein side chain conformational heterogeneity
- Concordance of X-ray and AlphaFold2 models of SARS-CoV-2 main protease with residual dipolar couplings measured in solution
- With the ICLR 2022 reviews released, there were quite a few protein papers:
A total of 10 submissions with "protein" or "antibody" in the title. 🧵
— Stephen Ra (@stephenrra) November 9, 2021
- Geometric transformers for protein interface contact prediction: https://t.co/rcgfwuuWc4
- Contrastive representation learning for 3d protein structures: https://t.co/Y8yORFTVGy
1/ https://t.co/Gb3hiuUN5D