
SCoV2-MD: a database for the dynamics of the SARS-CoV-2 proteome and variant impact predictions
Need an overview over the structural landscape of SARS-CoV-2? The database presented in this paper might be for you. Various models and experimental structures of protomers and oligomers in SARS-CoV-2 are presented together with a prediction of substrate and allosteric binding sites, protein-ligand docking, SARS-CoV-2 protein interactions with human proteins and impacts of mutations on stability mapped to their respective structures.
Two-input protein logic gate for computation in living cells
The authors design a “nano-computing agent”, a protein kinase that functions as a 2-bit logic OR gate. The first input, rapamycin, activates kinase activity and the second input, blue light, inactivates kinase activity. Analogous to hardware engineering complexities, inactivation wasn’t as fast as activation because of biological reasons. Similar setups could be applied to different kinds of proteins, giving us an arsenal of controllable protein agents for use in biomedicine and biotech.
Toward More General Embeddings for Protein Design: Harnessing Joint Representations of Sequence and Structure
In Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences, a transformer was trained on a huge dataset of sequences to embed proteins into a biologically meaningful learned embedding space. Here, the authors use this pre-trained model to generate sequence features, combine them with a graph representation of structures, and train a rotation and translation equivariant transformer to generate an embedding. This embedding performs better at mutation thermostability prediction likely because it’s able to take into account sequentially-distant structurally-close influences. Of course this structural information can come from computationally predicted structures as well, following the trend in this post-AlphaFold era of replacing or augmenting sequence-based approaches with structure.
Figure of the Week
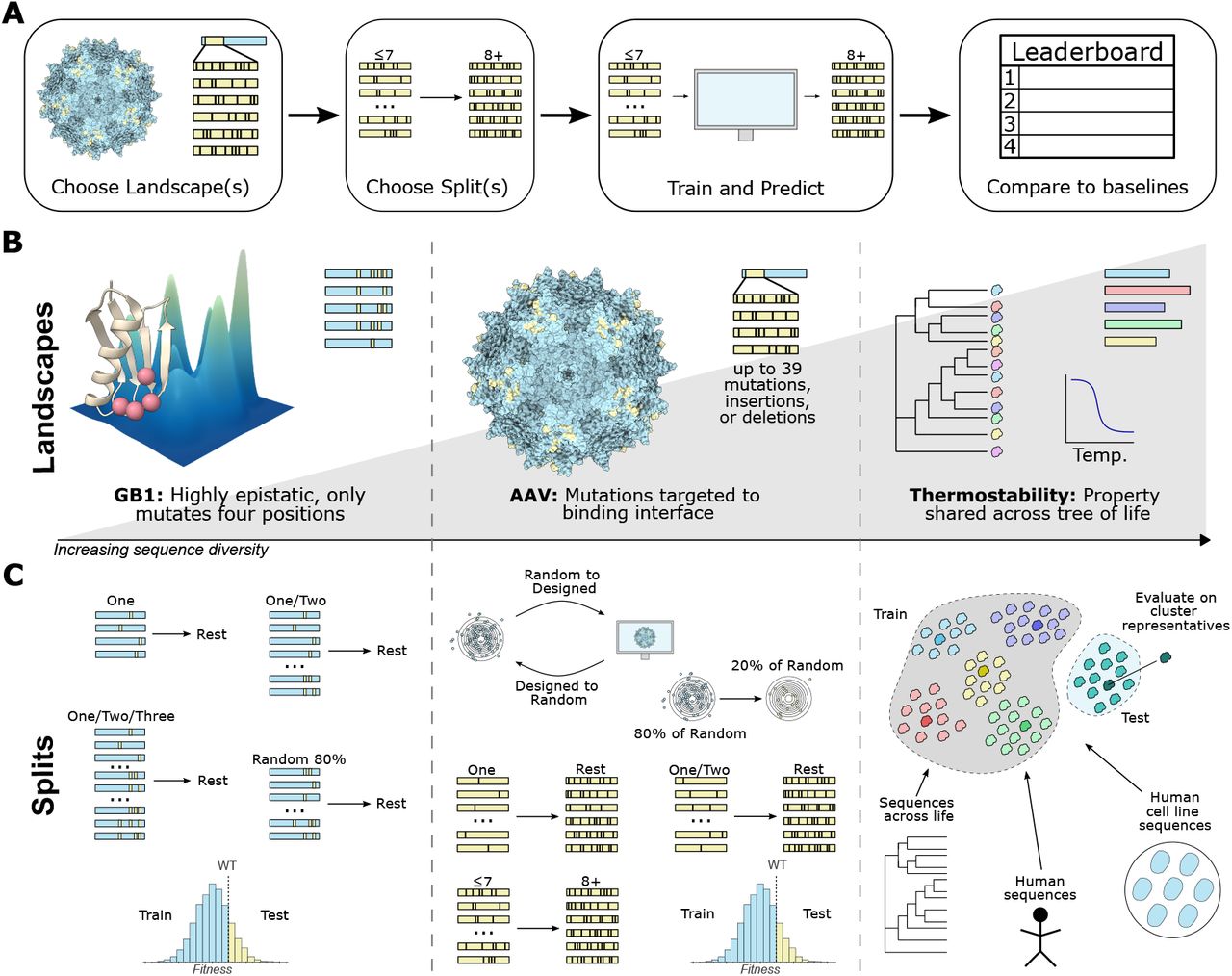
Figure 1: Summary of the workflow, landscapes, and splits. (A) General FLIP workflow: choose landscapes and splits that match user needs, train models and make predictions on the test set, and then compare to baseline models. (B) We choose landscapes that cover different types of sequence diversity. The GB1 landscape focuses on simultaneous mutation of four epistatic sites with nearly complete coverage [41] (PDB ID: 2GI9 [42]). The AAV capsid protein landscape sparsely samples sequences with up to 28 mutations, including insertions and deletions, to the the binding interface [43] (PDB ID: 6IH9 [44]). The thermostability landscape [45] measures a property shared by proteins from multiple functional groups across different domains of life. (C). We also provide up to seven suggested data splits for each landscape, which are described in Section 3.
Source: FLIP: Benchmark tasks in fitness landscape inference for proteins
Some more
- Computational design of nanoscale rotational mechanics in de novo protein assemblies
- Science Forum: Nanoscape, a data-driven 3D real-time interactive virtual cell environment
- FLIP: Benchmark tasks in fitness landscape inference for proteins
- Deep learning methods for designing proteins scaffolding functional sites
- ParaFold: Paralleling AlphaFold for Large-Scale Predictions
- #COVIDisAirborne: AI-Enabled Multiscale Computational Microscopy of Delta SARS-CoV-2 in a Respiratory Aerosol
- Computational Structural Genomics Unravels Common Folds and Novel Families in the Secretome of Fungal Phytopathogen Magnaporthe oryzae
- Contrastive learning on protein embeddings enlightens midnight zone at lightning speed
- Computational redesign of a fluorogen activating protein with Rosetta
- Multi-Scale Representation Learning on Proteins