
Molecular insights into receptor binding energetics and neutralization of SARS-CoV-2 variants
One of the most relevant protein complexes in the last two years is without a doubt the complex between the SARS-CoV-2 receptor binding domain (RBD) in the spike protein and the human ACE2 protein. This paper presents a quantitative analysis of the structural properties of the complex using atomic force microscopy and molecular dynamics. They observe that variants of concern tend to stabilize the RBD-ACE2 complex. The mutations N501Y (α, β, and γ strain) and E484Q (κ strain) particularly increase complex stability. N501Y is also shown to not affect antibody neutralization in a significant way. This analysis workflow might be generally applicable for the analysis of the impact of future viral mutations on infection-induced immunity.
Multi-State Modeling of G-protein Coupled Receptors at Experimental Accuracy
The authors observed the propensity of AlphaFold to model the inactive conformation of the members of the GPCR family. Presumably, this happens because of training based on the PDB where inactive state GPCR structures are overrepresented. However, they could successfully model the active state of the structures by using as input only templates from state-annotated GPCR databases and, surprisingly, avoiding the use of MSA input features. This work shows how it is possible to induce AlphaFold to model a specific conformation of a structure as long as experimental structures in multiple states are available to form state-specific template databases.
AlphaFill: enriching the AlphaFold models with ligands and co-factors
Already at the time of the CASP experiment, it was noted that even without the knowledge about ligands, especially metals, AlphaFold was able to accurately predict the right side chain orientation of ligand binding sites. Now that more than 300,000 structural models across 21 proteomes are available, that becomes even more evident, with AlphaFold predicting confidently even the structure of those proteins that require smaller molecules for folding. Missing from the models, however, are those ligands; AlphaFold does not predict (let’s say, yet) ligands. What AlphaFill provide is a glimpse into those ligands. By looking for close homologs of known structure with a ligand bound, the AlphaFill service (available as a database), “fills” the ligand-binding sites of AlphaFold models with those from the homologs. Neat and simple, the AlphaFill database provides a quite unique and super informative look into the structural coverage provided by the AlphaFold database.
Figure of the Week
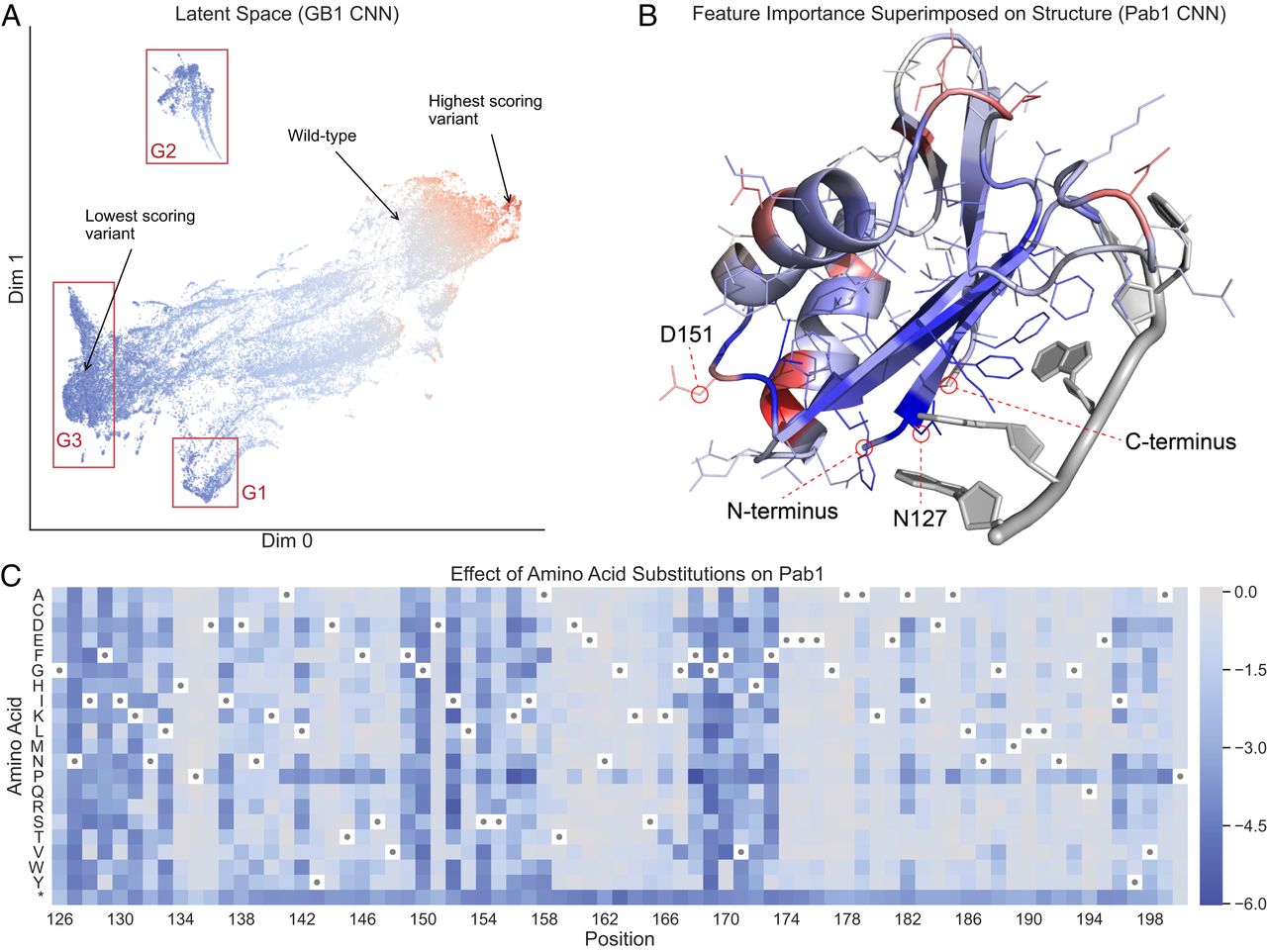
Fig 4. Neural network interpretation. (A) A UMAP projection of the latent space of the GB1 sequence convolutional network (CNN), as captured at the last internal layer of the network. In this latent space, similar variants are grouped together based on the transformation applied by the network to predict the functional score. Variants are colored by their true functional score, where red represents high-scoring variants and blue represents low-scoring variants. The clusters marked G1 and G2 correspond to variants with mutations at core residues near the start and end of the sequence, respectively. Cluster G3 corresponds to variants with mutations at surface interface residues. (B) Integrated gradients feature importance values for the Pab1 CNN, aggregated at each sequence position and superimposed on the protein’s three-dimensional structure. Blue represents positions with negative attributions, meaning mutations in those positions push the network to output lower scores, and red represents positions with positive attributions. (C) A heat map showing predictions for all single mutations from the Pab1 CNN. Wild-type residues are indicated with dots, and the asterisk is the stop codon. Most single mutations are predicted to be neutral or deleterious.
Source: Neural networks to learn protein sequence–function relationships from deep mutational scanning data
Some more
- De novo protein design by deep network hallucination
- Real-Time Structure Search and Structure Classification for AlphaFold Protein Models
- Loss-of-function, gain-of-function and dominant-negative mutations have profoundly different effects on protein structure: implications for variant effect prediction
- MUfoldQA_G: High-accuracy protein model QA via retraining and transformation
- Neural networks to learn protein sequence–function relationships from deep mutational scanning data
- Quantifying the binding landscapes of protein–protein interactions
- AlphaFold Models Illuminate Half of Dark Human Proteins
- EnzyHTP: A High-Throughput Computational Platform for Enzyme Modeling
- Protein loop modeling and refinement using deep learning models
- Ten things I ‘hate’ about refinement
- How Communication Pathways Bridge Local and Global Conformations in an IgG4 Antibody: a Molecular Dynamics Study