
Graphsite: ligand-binding site classification using deep graph neural network
The paper presents a graph convolutional neural network able to classify ligand-binding sites. The network takes as input a graph representing a protein pocket - the nodes represent atoms to which a few geometrical and chemical features are associated. After the convolution, the graph readout is performed with a very interesting graph pooling method based on self-attention: Set2Set. The network was trained on a dataset generated by the authors - they divided pockets into 14 different classes by clustering them according to the Tanimoto coefficient of the corresponding ligand. They validated the method by randomly splitting the dataset into a training and a testing set multiple times and averaging the obtained accuracies. Overall, they obtained a good prediction accuracy, but it would be interesting to observe the tool performances on an ‘external’ dataset.
Modeling Alternate Conformations with Alphafold2 via Modification of the Multiple Sequence Alignment
Yet another approach to coerce different conformations from AlphaFold2. We’ve covered two others just in the past two weeks. Even Deepmind ran into this for an elusive multi-drug transporter (LmrP) where vanilla AF2 only yielded one conformation - they pared the MSA and templates to only include structures with the other conformation and submitted the alternate conformations produced to CASP14. This paper builds on this idea in a novel way and gives us a neat procedure that can be more generally applied - make an AF2 model, pinpoint possible contact points (either from examining other crystal structures, prior knowledge from mutation studies, or just picking residues connecting two domains), then in-silico mutate the entire MSA at the chosen positions to alanine so that the attention network inside AF2, which depends on co-evolutionary signal, is forced to look elsewhere.
Navigating Chemical Space by Interfacing Generative Artificial Intelligence and Molecular Docking
A protein structure-aware molecular library preparation approach! A generative variational autoencoder is trained on molecular structures to obtain a latent space. Starting from an initial library, each reference molecule is embedded into this pretrained latent space and new molecules are selected from the neighborhood. These new molecules are docked to a target protein and those with the highest predicted binding affinity are chosen as the new set of reference molecules, and the process continues to narrow down the library to high affinity binders. There’s interactive TMAP plots covering some of their example runs - a really nice way of visualizing the greedy algorithm’s progress.
Figure of the Week
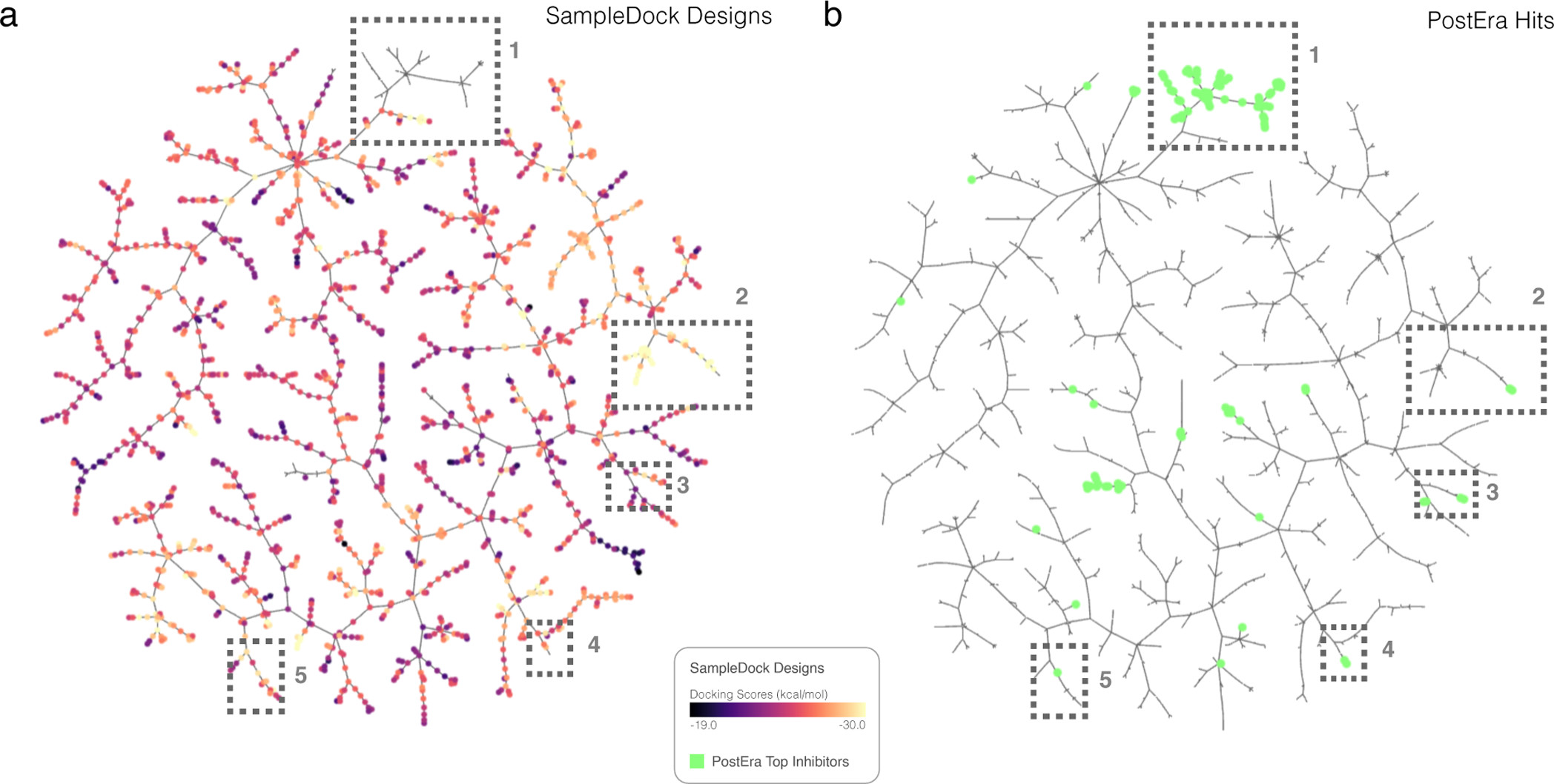
Figure 8. TMAP of best designs against the Mpro of SAR-CoV-2 target. The TMAPs with (a) our sample-and-dock designs and (b) the top-100 PostEra hits (bright green points) (https://covid.postera.ai/covid/activity_data) are shown. The sample-and-dock designs are color-coded based on docking scores. The corresponding interactive TMAP can be accessed via https://atfrank.github.io/SampleDock/vis_maps/tmap_mpro.html. The boxes highlight regions in the TMAP where sample-and-dock designs with low docking scores are near PostEra hits.
Source: Navigating Chemical Space by Interfacing Generative Artificial Intelligence and Molecular Docking
Some more
- De novo protein fold families expand the designable ligand binding site space
- DeepRank: a deep learning framework for data mining 3D protein-protein interfaces
- findMySequence: a neural-network-based approach for identification of unknown proteins in X-ray crystallography and cryo-EM
- Protein Structure Dynamic Prediction: a Machine Learning/Molecular Dynamic Approach to Investigate the Protein Conformational Sampling
- Exploiting Structural Modelling Tools to Explore Host-Translocated Effector Proteins
- De novo metalloprotein design
- A convolutional neural network highlights mutations relevant to antimicrobial resistance in Mycobacterium tuberculosis
- Cryptic Pockets Repository through Pocket Dynamics Tracking and Metadynamics on Essential Dynamics Space: Applications to Mcl-1
- Protein domain-based prediction of drug/compound–target interactions and experimental validation on LIM kinases
- AlphaFold and the amyloid landscape
- Hidden structure in disordered proteins is adaptive to intracellular changes
- Using Topology to Estimate Structural Similarities of Proteins
- Open-source protein docking in Rust!
LightDock, the open-source protein docking framework, has now been imported to Rust for improved speed.https://t.co/5sx5c51Jv7
— Nature Protocols (@NatureProtocols) December 6, 2021