
Leveraging nonstructural data to predict structures and affinities of protein–ligand complexes
The authors use the knowledge that even dissimilar ligands tend to bind to a given target protein in similar poses to improve computational binding pose and binding affinity prediction. Given a query ligand and target protein, ComBind finds other distinct ligands binding to that protein, docks all of them, and then picks poses for each ligand optimizing a potential shared across all the ligands, i.e. one that rewards protein-ligand interactions shared across different ligands. The same potentials are also used as a proxy for relative binding affinities, perfect for virtual screening. Quite a nice discussion comparing this to previous work and on integrating this concept with future improvements in docking and virtual screening.
Predictive profiling of SARS-CoV-2 variants by deep mutational learning
The emergence of SARS-CoV 2 variants with multiple, combined mutations rises the concern that the virus may fall into an evolutionary path that promotes the escaping of multiple classes of neutralizing antibodies. In this paper, the authors combined yeast display with deep sequencing to interrogate a massive sequence space of combinatorial mutations, representing billions of variants of the receptor binding motif of the SARS-CoV2 Spike protein. This allowed them to generate a large, experimental labelled dataset of sequences that could then be used to train two machine learning models (a random forest and a long-short-term-memory recurrent neural network) that predict, at very high accuracy, whether a given variant would interact with the ACE2 enzyme and, if so, would escape four commercial neutralizing antibodies. The models may enable researchers to select candidate antibody therapeutics and could be used as a de facto monitoring system, helping researchers to prospectively identify future variants with the highest likelihood of emergence.
Conformational variation in enzyme catalysis: A structural study on catalytic residues
A very thorough analysis of the flexibility of catalytic residues from the Mechanism and Catalytic Site Atlas across over 900 enzyme families and across the entire PDB. Some conclusions: there’s a high structural conservation of the active site across enzymes from the same family almost regardless of the sequence similarity; most active sites exhibit some degree of flexibility, half of which is accounted for by only side chain motion; the highest RMSD variability is observed when comparing ligand-free and ligand-bound active sites; “reactant” residues (proton donor, acceptor, and shuttle) are more rigid than “spectator” (electrostatic stabiliser, activator, steric role) residues; and, based on their RMSD distributions, active sites can be broadly divided into inherently rigid, inherently flexible, open/closed, and extensively variable. Look out for their CSA-3D Python library which will help generalize analyses on catalytic sites like this one.
Figure of the Week
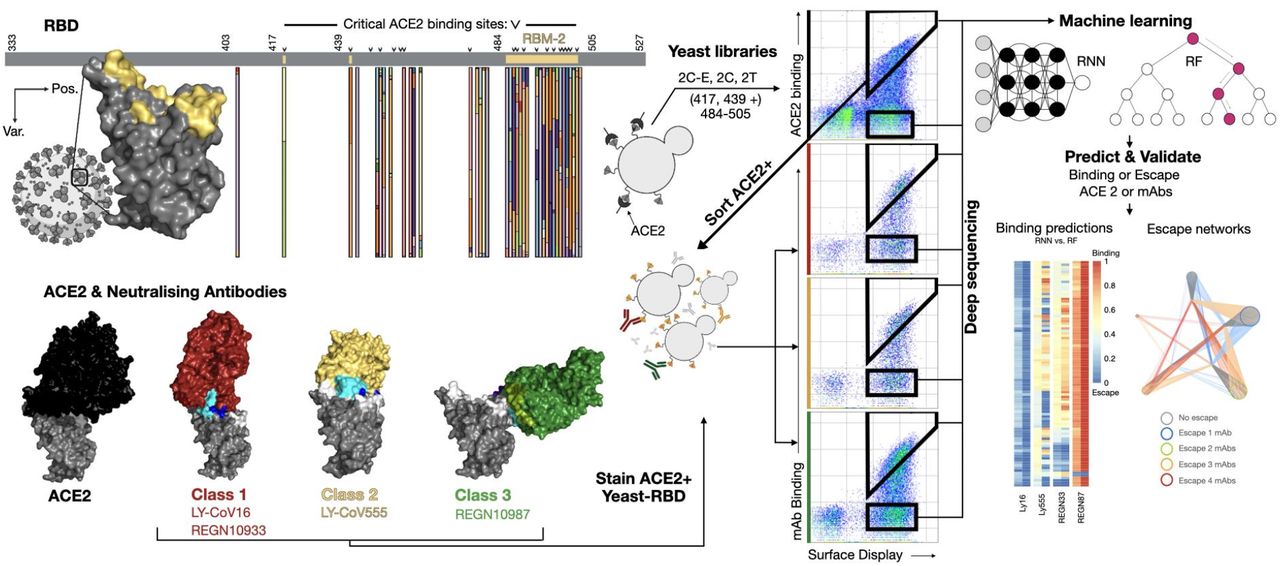
Figure 1. Overview of deep mutational learning of the RBD for prediction of ACE2 binding and antibody escape. The RBD or the SARS-CoV-2 spike protein is expressed on the surface of yeast, mutagenesis libraries are designed on the receptor-binding motif (RBM-2) of the RBD, which are the sites of interaction with ACE2 and neutralizing antibodies (e.g., therapeutic antibody drugs). RBD libraries are screened by FACS for binding to ACE2 and neutralizing antibodies, both binding and non-binding (escape) populations are isolated and subjected to deep sequencing. Machine learning models are trained to predict binding status to ACE2 or antibodies based on RBD sequence. Machine learning models are then used to predict ACE2 binding and antibody escape on current and prospective variants and lineages.
Source: Predictive profiling of SARS-CoV-2 variants by deep mutational learning
Some more
- SARS-CoV-2 variants impact RBD conformational dynamics and ACE2 accessibility
- Synthon-based ligand discovery in virtual libraries of over 11 billion compounds
- Learning physics confers pose-sensitivity in structure-based virtual screening
- InDeep: 3D fully convolutional neural networks to assist in silico drug design on protein-protein interactions
- A novel sequence-based predictor for identifying and characterizing thermophilic proteins using estimated propensity scores of dipeptides
- Constructing synthetic-protein assemblies from de novo designed 310 helices
- Version 3 of Improved prediction of protein-protein interactions using AlphaFold2
… “In short two major things have changed 1) We have introduced a pDockQ score that predicts the DockQ score for each protein-protein interactions. This improves both separation of good and bad models and interacting and non-interacting proteins. 2) We have added comparisons to (a) state of the art docking method and (b) alphafold-multimer.”
- Version 2 of Applications of AlphaFold beyond Protein Structure Prediction
- Don’t forget to check out the papers from the Machine Learning in Structural Biology workshop!