
PDBspheres - a method for finding 3D similarities in local regions in proteins
The authors collect an impressive library of over 2.6 million “spheres” cropped from ligand-binding and peptide-binding sites in the PDB. This library, typically filtered based on the ligand of interest, ligand size, or sequence identity to the protein of interest, serves as a search database for finding pockets in a query protein, with the filtering reducing the search time down from days to hours. The search itself depends on a structure alignment of the sphere to the entire query protein, returning multiple detected pockets and improving upon pocket detection performance state-of-the-art even at low sequence identities. Surprisingly, side-chain conformations do not improve performance. In another experiment, the authors also show that binding affinity is similar across similar pockets.
Improving AlphaFold modeling using implicit information from experimental density maps
In this paper the authors bring AlphaFold into the integrative modelling and iterative model building realms, proposing a new approach for protein model building starting from its sequence and experimental cryo-EM map. From an initial AlphaFold2 model, the authors propose an iterative approach where that model is fit into the density, remodelled based on it, and fed back as a template for another round of AlphaFold2 structure prediction. They applied the approach to the modelling of 25 cryo-EM structures and, in most cases, the procedure allowed for the building of models at a better accuracy than the initial AlphaFold model. While here they combine AlphaFold models with cryo-EM map interpretation, the approach is general and can be extended to any other protein structure prediction method and map-based biophysical approach, thus paving the way to the development of further synergic methods for better modelling of protein structures at atomic level of accuracy.
AlphaFold2-aware protein-DNA binding site prediction using graph transformer
The authors present GraphSite, a graph-based predictor of DNA binding sites at a residue level. This tool is based on a graph representation of the protein in which the residues are the nodes, and uses a transformer architecture to classify them. The nodes features are generated using both multiple sequence alignment and structural information. To do so, they predict the structure with AlphaFold-2 and extract the intermediate representation generated by the network. Moreover, they extract several structural properties from the predicted model such as backbone angles and accessible surface area. This method offers very good classification performances compared both to the sequence-based and structure-based state-of-the-art methods. Unsurprisingly, predicted structure quality can greatly affect performance. The authors also mention an interesting possible extension of their approach: adding edge features encoding residue distance or AlphaFold2 intermediate pair representations.
Figure of the Week
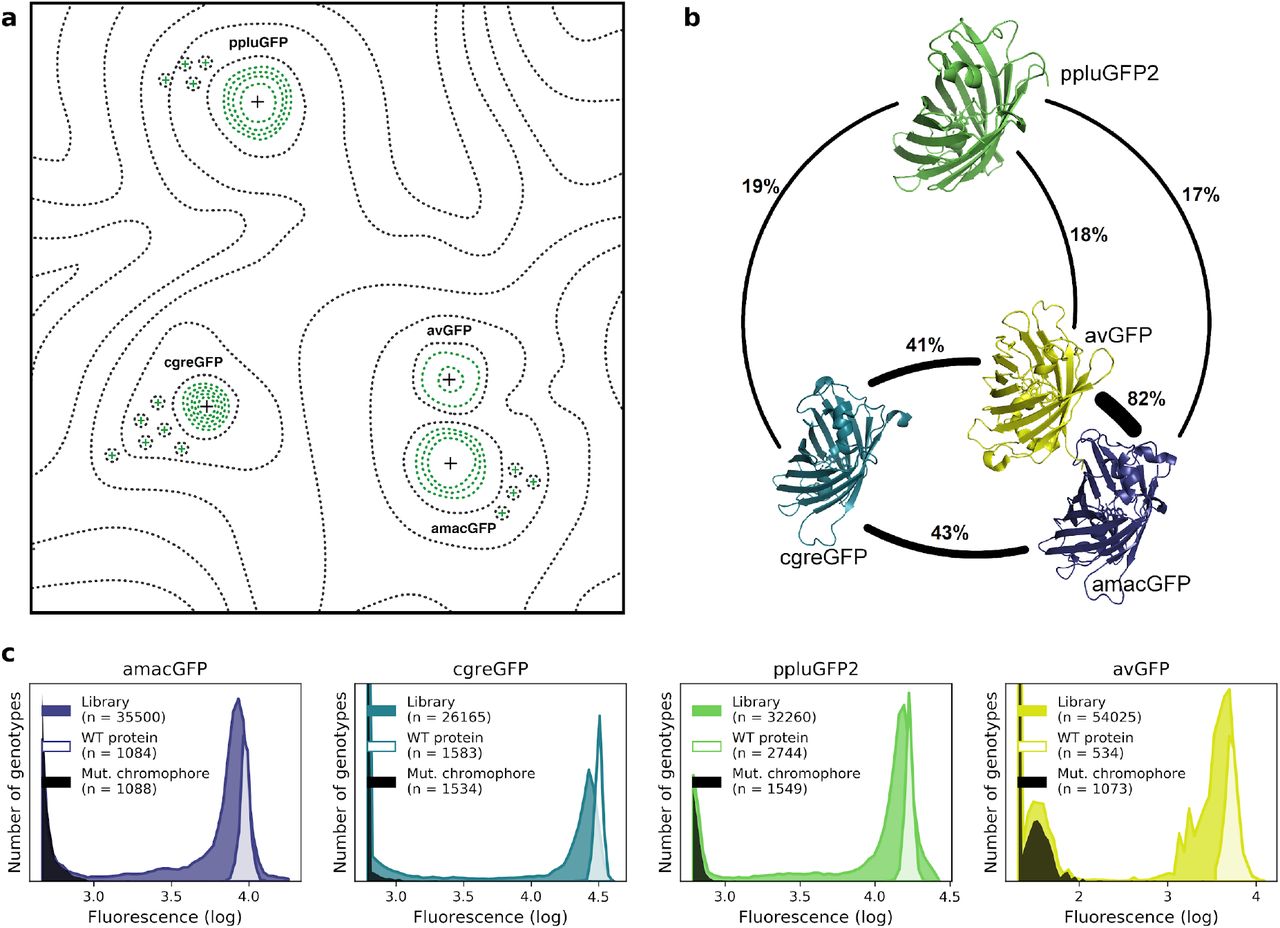
Figure 1. Comparison of four GFP fitness peaks. a, A conceptual representation of the GFP fitness landscape following the visualization proposed by Sewal Wright (Wright, 1932). The black dotted lines represent the unknown regions of the fitness landscape and the green lines the surveyed local fitness peak. Wildtype GFPs (black +) and predicted functional GFPs (green +) are shown at an approximate scale of sequence divergence from each other. b, Amino acid sequence identity between different orthologs, displayed in percent. c, Distribution of fluorescence of mutant libraries (colour), control wildtype protein sequences (white), and protein sequences containing loss-of-function mutations in the chromophore (black).
Source: Heterogeneity of the GFP fitness landscape and data-driven protein design
Some more
- Using metagenomic data to boost protein structure prediction and discovery
- Heterogeneity of the GFP fitness landscape and data-driven protein design
- The new version of Phenix lets you carry out the kinds of integrative modelling described above
- A new Foldseek server is online:
Search your protein structures against #AlphaFold DBs and #PDB in seconds using our Foldseek server. Just paste your PDB file and click search. We offer local (SW) and global (TMalign) structural alignments. Server was build by the amazing @milot_mirdita
— Martin Steinegger 🇺🇦 (@thesteinegger) January 3, 2022
🚀https://t.co/yZoYA0iTJz pic.twitter.com/bvarZye0XY