Sequence-structure-function relationships in the microbial protein universe
Metagenomics is a big source of novelties, with many genes coding for proteins without any clear homology to well-known families. In this work, Leman et al. modelled ~200,000 structures for diverse protein sequences without clear homologs from 1,003 representative metagenomes. This work was initiated 5 years ago, before the AlphaFold era, and modelling was carried out using Rosetta and DMPfold through a large-scale citizen science initiative, the World Community Grid project. What they found when putting all these structures in context (using a graphlet-based network embedding GRAFENE) is that, despite most of these proteins representing seemingly orphan genes, they adopt already known folds, with only ~160 novel folds uncovered, suggesting that structural space is saturated. They further predicted functions using DeepFri and unravelled various cases where the same function is mapped to unrelated folds, highlighting parallel, possibly backup functional routes. All together, this work demonstrates how large-scale protein sequence-structure-function predictions can highlight the origin and importance of seemingly metagenomic novelties.
An end-to-end deep learning method for rotamer-free protein side-chain packing
Goodbye rotamer libraries? For decades they were the undisputed starting point for any side-chain reconstruction problem. Given a discrete set of possible side-chain conformations at each location, Monte Carlo or graph based algorithms provided solutions optimizing some pairwise energy function. Here, the authors present AttnPacker. AttnPacker uses spatial information derived from protein backbone coordinates to efficiently model residue and pairwise neighborhoods. This, coupled with an SE(3)-equivariant architecture, allows for the simultaneous prediction of all amino acid side-chain coordinates without the use of rotamer libraries or conformational sampling. Yes, it’s fast, yes it’s accurate.
Disentanglement of Entropy and Coevolution using Spectral Regularization
Deep learning methods for protein structure prediction and other tasks often make heavy use of coevolutionary signal. These typically use statistical models to generate a matrix (modelling conservation and entropy) and a 4D tensor (modeling the coevolution). In tasks like contact prediction, the tensor is often also reduced to a matrix, representing the strength of residue-residue interactions in a protein. However, this matrix and the original tensor are highly correlated with entropy, remedied by performing a low-rank correction as a post-processing step. Approaches that use the tensor directly or don’t perform this correction may struggle to disentangle entropy from coevolution. The authors here propose a novel spectral regularizer that works by removing the dominant eigenmode of the coevolution matrix, thereby sparsifying the structural information and removing entropic correlation. They test their approach on contact prediction and sequence design tasks, and compete favourably with approaches requiring the post-processing step. They also show that their regularizer is interpretable, revealing attractive and repulsive interactions. Something like this will likely make its way into future structure prediction methods, to improve prediction as well as the learned hidden representations.
Figure of the Week
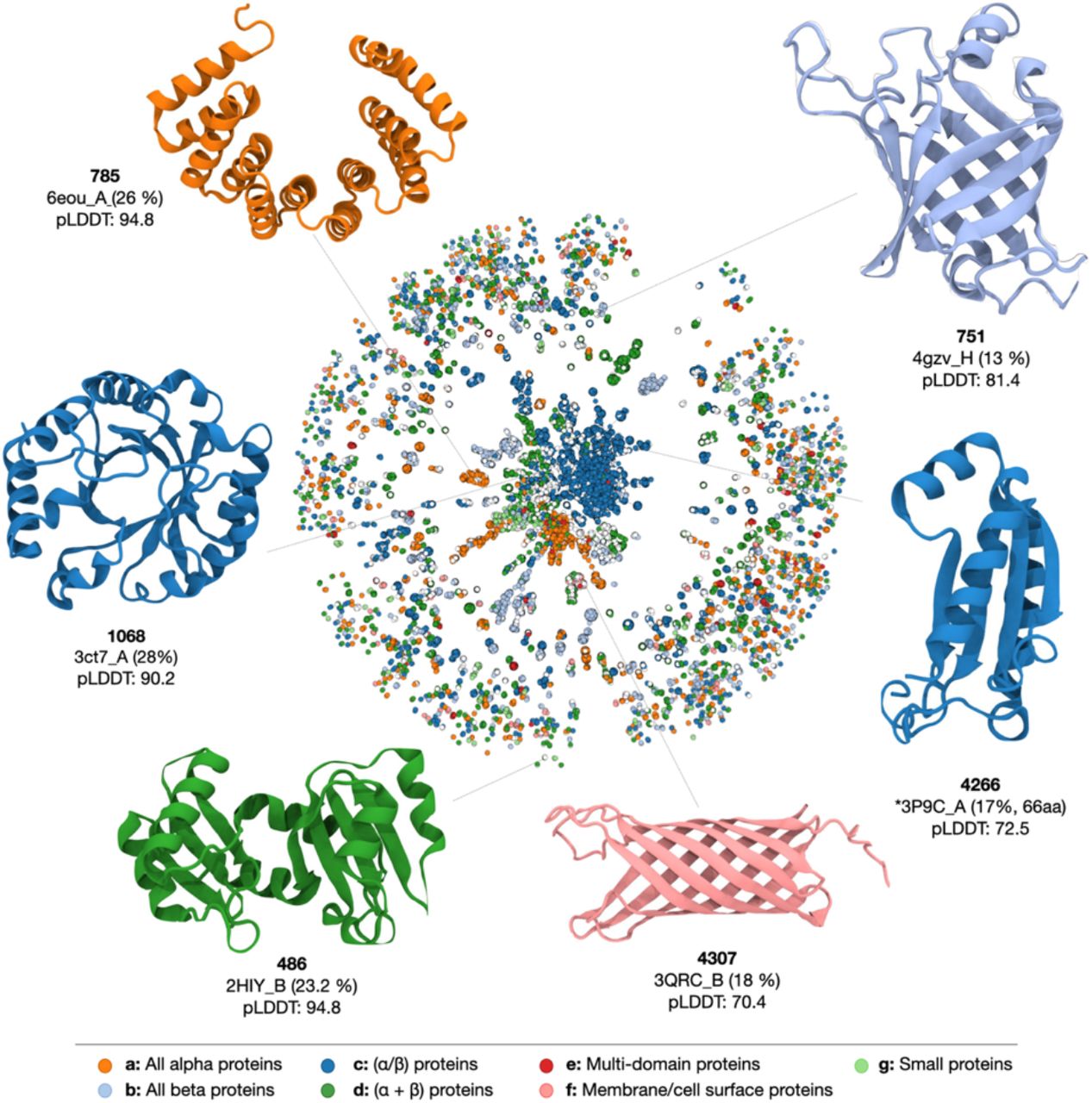
Figure 3: An overview of the protein space and examples of proteins generated by ProtGPT2. Each node represents a sequence. Two nodes are linked when they have an alignment of at least 20 amino acids and 70 % HHsearch probability. Colours depict the different SCOPe classes, and ProtGPT2 sequences are shown in white. As examples, we select proteins of each of the major 5 SCOP classes: all-β structures (751), α/β (4266, 1068), membrane protein (4307), α+β (486), and all-α (785). The selected structures are colored according to the class of their most similar hit. The structures were predicted with AlphaFold, and we indicate the code of the most similar structure in the PDB as found by FoldSeek (35), except for protein 4266, where no structures were found.
Source: A deep unsupervised language model for protein design
Some more
- Deep learning in prediction of intrinsic disorder in proteins
- State-of-the-Art Estimation of Protein Model Accuracy using AlphaFold
- Predicting the structure of large protein complexes using AlphaFold and sequential assembly
- AlphaFold Models of Small Proteins Rival the Accuracy of Solution NMR Structures
- AlphaFold2 fails to predict protein fold switching
- Accurate determination of protein:ligand standard binding free energies from molecular dynamics simulations
- Protein Structure Representation Learning by Geometric Pretraining
- Modeling the Dynamics of Protein–Protein Interfaces, How and Why?
- Oncological drug discovery: AI meets structure-based computational research
- Decomposing compounds enables reconstruction of interaction fingerprints for structure-based drug screening
- A deep unsupervised language model for protein design
- Improving protein–ligand docking and screening accuracies by incorporating a scoring function correction term
- Interleukin-2 superkines by computational design
- AlphaFold encodes the principles to identify high affinity peptide binders
- DENVIS: scalable and high-throughput virtual screening using graph neural networks with atomic and surface protein pocket features
- Accurate Protein Domain Structure Annotation with DomainMapper
- Fragment libraries designed to be functionally diverse recover protein binding information more efficiently than standard structurally diverse libraries